ECLIP data analysis using CTK
From Zhang Laboratory
Contents
Introduction
This tutorial outlines how to analyze CLIP data derived from the enhanced CLIP (eCLIP) protocol. eCLIP incorporates modifications of the iCLIP protocol. Instead of utilizing circular ligation, the eCLIP protocol uses ligation of two separate adapters (an indexed 3' RNA adapter that is ligated to the crosslinked RNA fragment while on immunoprecipitation beads and and 3' ssDNA adapter that is ligated after reverse transcription) added in two separate steps. For more information on the biochemical iCLIP protocol, refer to the following reference:
Van Nostrand, E. L., G. A. Pratt, A. A. Shishkin, C. Gelboin-Burkhart, M. Y. Fang, B. Sundararaman, S. M. Blue, T. B. Nguyen, C. Surka, K. Elkins, R. Stanton, F. Rigo, M. Guttman and G. W. Yeo (2016). "Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP)." Nat Methods 13(6): 508-514.
eCLIP reads typically have the following structure (read 2, which corresponds to the sense strand):
NNNNN[CLIP tag sequence]SSSSSSSAGATCGGAAGAGCGTCGTGT
where N5 is the random barcode (UMI). In some libraries this could be 10 nt (N10).
S7 is the sample index, which at least the following possible 7mers: ATTGCTT (A01), ACAAGCC (B06), AACTTGT (C01), AGGACCA (D08). The sample index could also include 4 extra degenerate nucleotides, and takes the following form: ANNNNGGTCAT (A03), ANNNNACAGGA (G07), ANNNNAAGCTG (A04), ANNNNGTATCC (F05).
Sample dataset
We will use RBFOX2 eCLIP data from HepG2 cells for this tutorial, that were generated as part of the ENCODE project.
Van Nostrand, E. L., G. A. Pratt, A. A. Shishkin, C. Gelboin-Burkhart, M. Y. Fang, B. Sundararaman, S. M. Blue, T. B. Nguyen, C. Surka, K. Elkins, R. Stanton, F. Rigo, M. Guttman and G. W. Yeo (2016). "Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP)." Nat Methods 13(6): 508-514.
Raw FASTQ files
We will use ~/test as our working directory, and download fastq files under ~/test/fastq
mkdir ~/test; cd ~/test mkdir fastq; cd fastq
The raw sequence files from Illumina sequencing can be downloaded from the ENCODE web site.
wget https://www.encodeproject.org/files/ENCFF647KDW/@@download/ENCFF647KDW.fastq.gz wget https://www.encodeproject.org/files/ENCFF289OFA/@@download/ENCFF289OFA.fastq.gz #link files with more meaningful names ln -s ENCFF647KDW.fastq.gz HepG2.RBFOX2.rep1.R2.fastq.gz ln -s ENCFF289OFA.fastq.gz HepG2.RBFOX2.rep2.R2.fastq.gz
Note that the read2 correspond to the sense strand. Following the original study (Van Nostrand et al. 2016), only read2 is used for analysis in this tutorial.
Output files of major steps generated in this tutorial
- Fully preprocessed FASTQ files (489 MB) : output files generated for use right before mapping.
- Unique CLIP tag and mutation files (424 MB) : output files generated after mapping and collapsing PCR duplicates.
- Peak calling files (25 MB) : output files after peak calling.
- CIMS output files (1.5 MB) : output files after CIMS analysis.
- CITS output files (587 KB) : output files after CITS analysis.
The number of reads in each file, after each step of processing, is summarized in Table 1 below.
| Protein | Sample | # of raw reads | # of trimmed & filtered reads | # of collapsed reads | # of mapped reads | # of reads after removal of repetitive RNAs | # of unique tags |
|---|---|---|---|---|---|---|---|
| RBFOX2 | rep1 | 10,038,858 | 9,907,629 | 7,652,372 | 4,809,641 | 4,498,170 | 4,094,510 |
| RBFOX2 | rep2 | 10,236,928 | 10,066,151 | 7,462,881 | 4,788,594 | 4,405,465 | 4,085,826 |
| Total | 20,275,786 | 19,973,780 | 15,115,253 | 9,598,235 | 8,903,635 | 8,180,336 |
Read preprocessing
Demultiplexing samples
The files downloaded from SRA have already be demultiplexed. Therefore this step has already been performed.
For the user who would like to start from a FASTQ file consisting of multiple libraries, check here.
In addition, in the demultiplexed fastq files, the random barcode has already been moved and attached to the begin of the read ID.
The barcode length can be either 5 nt or 10 nt, and it is important for this to be determined by checking a few reads, e.g.,
@CCAAC:SN1001:449:HGTN3ADXX:1:1101:1373:1964 2:N:0:1 GAGAGAGGAGTGGGAAGTTGGGATAGTACCCAGAGAGAGAGGCCCG + FFFFFBFFBFBFFFFFIFFFIFFIFIIIIIIFIIIIFFIFIIFFIF
Both replicates of RBFOX2 eCLIP have a barcode length of 5 nt. To be compatible with the CTK package, we reformatted the fastq files by moving the random barcode back to the beginning of each read (using a script not included in the CTK package as this is not a standard step of CLIP data analysis from raw data). The read illustrated above will become:
@SN1001:449:HGTN3ADXX:1:1101:1373:1964 2:N:0:1 CCAACGAGAGAGGAGTGGGAAGTTGGGATAGTACCCAGAGAGAGAGGCCCG + IIIIIFFFFFBFFBFBFFFFFIFFFIFFIFIIIIIIFIIIIFFIFIIFFIF
Note the quality score line needs to be modified accordingly (5 I's were added at the front in the example above).
Read quality filtering
For this particular dataset, we skipped the read quality filtering step, as this does not appear to make any major impact on the results.
For the user who would like to include this step, check here.
Trimming of 3' linker sequences
cd ~/test mkdir filtering cd filtering
One can then trim the 3' adapter using the command below.
#!/bin/bash
for f in HepG2.RBFOX2.rep1.R2 HepG2.RBFOX2.rep2.R2; do
cutadapt -f fastq --match-read-wildcards --times 1 -e 0.1 -O 1 --quality-cutoff 5 -m 20 \
-a ATTGCTTAGATCGGAAGAGCGTCGTGT -a ACAAGCCAGATCGGAAGAGCGTCGTGT -a AACTTGTAGATCGGAAGAGCGTCGTGT -a AGGACCAAGATCGGAAGAGCGTCGTGT \
-a ANNNNGGTCATAGATCGGAAGAGCGTCGTGT -a ANNNNACAGGAAGATCGGAAGAGCGTCGTGT -a ANNNNAAGCTGAGATCGGAAGAGCGTCGTGT -a ANNNNGTATCCAGATCGGAAGAGCGTCGTGT \
-o $f.trim.fastq.gz ../fastq/$f.reformat.fastq.gz > $f.cutadpt.log
done
The parameters are as follows:
- -f fastq : input is a FASTQ file
- --match-read-wildcards : allow N in the read as matches
- --times 1 : removes up to 1 adapters from each read; here we specify 2 as occasionally some tags have 2 copies of adapters
- -e 0.1 : maximum allowed error rate is 0.1
- -O 1 : the read is not modified if the overlap between the read and the adapter is shorter than 1nt
- --quality-cutoff : refers to quality
- -m 20 : minimum length of the read being 24nt (5 nt barcode + 15 nt tags)
- -a : the 3' adapter
For more cutadapt usage information:
cutadapt --help
This is the same thing, but using a Sun Grid Engine to execute this UNIX job on a remote machine.
#!/bin/bash
#$ -t 1-2 -m a -cwd -N CLIP
files=(HepG2.RBFOX2.rep1.R2 HepG2.RBFOX2.rep2.R2);
f=${files[$SGE_TASK_ID-1]}
cutadapt -f fastq --match-read-wildcards --times 1 -e 0.1 -O 1 --quality-cutoff 5 -m 20 \
-a ATTGCTTAGATCGGAAGAGCGTCGTGT -a ACAAGCCAGATCGGAAGAGCGTCGTGT -a AACTTGTAGATCGGAAGAGCGTCGTGT -a AGGACCAAGATCGGAAGAGCGTCGTGT \
-a ANNNNGGTCATAGATCGGAAGAGCGTCGTGT -a ANNNNACAGGAAGATCGGAAGAGCGTCGTGT -a ANNNNAAGCTGAGATCGGAAGAGCGTCGTGT -a ANNNNGTATCCAGATCGGAAGAGCGTCGTGT \
-o $f.trim.fastq.gz ../fastq/$f.reformat.fastq.gz > $f.cutadpt.log
Note 1: We included all 8 version of possible 3' adaptors to be trimmed, as the exact adaptors used in the specific libraries were not documented.
Note 2: It is good to check the number of reads by running the following command:
#e.g., for raw reads
for f in `ls ../fastq/*.fastq.gz`; do
c=`zcat $f | wc -l`
c=$((c/4))
echo $f $c
done
#e.g., for trimmed reads
for f in `ls *.trim.fastq.gz`; do
c=`zcat $f | wc -l`
c=$((c/4))
echo $f $c
done
Collapse Exact PCR Duplicates
Collapse exact PCR duplicates such that if multiple reads have exactly the same sequence, only one is kept.
#!/bin/bash -l for f in HepG2.RBFOX2.rep1.R2 HepG2.RBFOX2.rep2.R2 HepG2.RBFOX2_mock_input.rep1.R2 do perl /usr/local/CTK/fastq2collapse.pl $f.trim.fastq.gz - | gzip -c > $f.trim.c.fastq.gz done
Note 1: It is good to check the number of reads by running the command:
for f in `ls *.trim.c.fastq.gz`; do
c=`zcat $f | wc -l`
c=$((c/4))
echo $f $c
done
Strip random barcode (UMI)
Remove the 5' degenerate barcode. For these particular libraries, the barcode length is 5 nt (it could be 10 nt for some of the other eCLIP libraries).
for f in HepG2.RBFOX2.rep1.R2 HepG2.RBFOX2.rep2.R2; do
perl /usr/local/CTK/stripBarcode.pl -format fastq -len 5 $f.trim.c.fastq.gz - | gzip -c > $f.trim.c.tag.fastq.gz
done
Note 1: Get the distribution of tag lengths for diagnostic purposes using the following command.
for f in HepG2.RBFOX2.rep1.R2 HepG2.RBFOX2.rep2.R2; do
zcat $f.trim.c.tag.fastq.gz | awk '{if(NR%4==2) {print length($0)}}' | sort -n | uniq -c | awk '{print $2"\t"$1}' > $f.trim.c.tag.seqlen.stat.txt
done
Download fully preprocessed FASTQ files here(489 MB).
Read mapping & parsing
We assume the reference index has been prepared and is available under /genomes/hg19/bwa/.
cd ~/test mkdir mapping cd mapping
Run bwa to align the reads to the reference genome.
for f in HepG2.RBFOX2.rep1.R2 HepG2.RBFOX2.rep2.R2; do bwa aln -t 4 -n 0.06 -q 20 /genomes/hg19/bwa/hg19.fa ../filtering/$f.trim.c.tag.fastq.gz > $f.sai bwa samse /genomes/hg19/bwa/hg19.fa $f.sai ../filtering/$f.trim.c.tag.fastq.gz | gzip -c > $f.sam.gz done
The option -n 0.06 specifies slightly more stringent criteria than the default. The number of allowed mismatches (substitutions or indels) depending on read length is as follows:
[bwa_aln] 17bp reads: max_diff = 1 [bwa_aln] 20bp reads: max_diff = 2 [bwa_aln] 45bp reads: max_diff = 3 [bwa_aln] 73bp reads: max_diff = 4 [bwa_aln] 104bp reads: max_diff = 5 [bwa_aln] 137bp reads: max_diff = 6 [bwa_aln] 172bp reads: max_diff = 7 [bwa_aln] 208bp reads: max_diff = 8 [bwa_aln] 244bp reads: max_diff = 9
The -q option is used to trim low quality reads (an average of 20 or below). However, this does not seem to do too much after the trimming steps above.
Parsing SAM file
for f in HepG2.RBFOX2.rep1.R2 HepG2.RBFOX2.rep2.R2; do perl /usr/local/CTK/parseAlignment.pl -v --map-qual 1 --min-len 18 --mutation-file $f.mutation.txt $f.sam.gz - | gzip -c > $f.tag.bed.gz done
This will keep only unique mappings (with MAPQ >=1) and a minimal mapping size of 18 nt.
Note 1: In the tag bed file, the 5th column records the number of mismatches (substitutions) in each read
Note 2: Other aligners might not use a positive MAPQ as an indication of unique mapping.
Another useful option is --indel-to-end, which specifies the number of nucleotides towards the end from which indels should not be called (default=5 nt).
Note 3: The parsing script relies on MD tags, which is an optional field without strict definition in SAM file format specification. Some aligners might have slightly different format how they report mismatches. If other aligners than bwa is used, one should run the following command:
samtools view -bS $f.sam | samtools sort - $f.sorted samtools fillmd $f.sorted.bam /genomes/hg19/bwa/hg19.fa | gzip -c > $f.sorted.md.sam.gz
This will ensure the sam file gets parsed properly.
Note 4: Keep track what proportion of reads can be mapped uniquely.
wc -l *.tag.bed
Remove tags from rRNA and other repetitive RNA (optional)
This is an optional step, and performed here following the original analysis of this dataset (see Van Nostrand et al. 2016 Nat Meth 13:508-514 for more detail). In some cases, CLIP data can have contaminants due to abundant repetitive RNAs (such as rRNAs).
for f in HepG2.RBFOX2.rep1.R2 HepG2.RBFOX2.rep2.R2; do perl /usr/local/CTK/tagoverlap.pl -big -region /usr/local/CTK/annotation/genomes/hg19/annotation/rmsk.RNA.bed -ss --complete-overlap -r --keep-tag-name --keep-score -v $f.tag.bed $f.tag.norRNA.bed done
As of now, the path to the BED file with a database of repetitive RNAs has to be specified directly, which is somewhat inconvenient. This will be improved in the future.
Collapse PCR duplicates
It is critical to collapse PCR duplicates, not only for the exact duplicates collapsed above, but also for those with slight differences due to sequencing errors. These sequencing errors can also occur in the random barcode, so a naive method to collapse the same barcode is frequently insufficient.
for f in HepG2.RBFOX2.rep1.R2 HepG2.RBFOX2.rep2.R2; do perl /usr/local/CTK/tag2collapse.pl -big -v --random-barcode -EM 30 --seq-error-model alignment -weight --weight-in-name --keep-max-score --keep-tag-name $f.tag.norRNA.bed $f.tag.uniq.bed done
In the command above, tags mapped to the same genomic positions (i.e., the same start coordinates for the 5' end of RNA tag) will be collapsed together if they share the same or "similar" barcodes. A model-based algorithm is used to identify "sufficiently distinct" barcodes. This algorithm was described in detail in the following paper:
Darnell JC, et al. FMRP Stalls Ribosomal Translocation on mRNAs Linked to Synaptic Function and Autism. Cell. 2011; 146:247–261.
Compared to the original algorithm, the current implementation estimates sequencing error from aligned reads, which is then fixed during the iterative EM procedure.
Note 1: Sequencing errors in the degenerate barcodes are estimated from results of read alignment. The number of substitutions in each read must be provided in the 5th column.
Note 2: Note that the read ID in the input BED file (in the 4th column) must take the form READ#x#NNNNN, where x is the number of exact duplicates and NNNNN is the bar-code nucleotide sequence appended to read IDs in previous steps. Read IDs that are not in this format will generate an error.
Note 3: As a diagnostic step, get the length distribution of unique tags, which should be a more faithful representation of the library:
for f in HepG2.RBFOX2.rep1.R2 HepG2.RBFOX2.rep2.R2; do
awk '{print $3-$2}' $f.tag.uniq.bed | sort -n | uniq -c | awk '{print $2"\t"$1}' > $f.tag.uniq.len.dist.txt
done
Get the mutations in unique tags.
for f in HepG2.RBFOX2.rep1.R2 HepG2.RBFOX2.rep2.R2; do python /usr/local/CTK/joinWrapper.py $f.mutation.txt $f.tag.uniq.bed 4 4 N $f.tag.uniq.mutation.txt done
This script is from galaxy, and included for your convenience. The parameters 4 4 indicate the columns in the two input file used to join the two, and N indicates that only paired rows should be printed.
Table 2 summarizes the number of unique mutations of different types in each sample.
| Protein | Sample | # of unique tags | Total # of mutations in unique tags | Deletions | Insertions | Substitutions |
|---|---|---|---|---|---|---|
| RBFOX2 | rep1 | 4,094,510 | 754,097 | 123,895 | 12,833 | 617,369 |
| RBFOX2 | rep2 | 4,085,826 | 616,218 | 72,627 | 11,418 | 532,173 |
| Total | 8,180,336 | 1,370,315 | 196,522 | 24,251 | 1,149,542 |
Merging biological replicates
After getting unique tags, one might want to concatenate these runs, which can be distinguished by different colors. As an example:
perl /usr/local/CTK/bed2rgb.pl -v -col "188,0,0" HepG2.RBFOX2.rep1.R2.tag.uniq.bed HepG2.RBFOX2.rep1.R2.tag.uniq.rgb.bed perl /usr/local/CTK/bed2rgb.pl -v -col "128,0,128" HepG2.RBFOX2.rep2.R2.tag.uniq.bed HepG2.RBFOX2.rep2.R2.tag.uniq.rgb.bed
Then these can be concatenated.
cat HepG2.RBFOX2.rep1.R2.tag.uniq.rgb.bed HepG2.RBFOX2.rep2.R2.tag.uniq.rgb.bed > HepG2.RBFOX2.pool.R2.tag.uniq.rgb.bed cat HepG2.RBFOX2.rep1.R2.tag.uniq.mutation.txt HepG2.RBFOX2.rep2.R2.tag.uniq.mutation.txt > HepG2.pool.R2.tag.uniq.mutation.txt
Download unique tag and mutation files here (424MB).
Annotating and visualizing CLIP tags
Get the genomic distribution of CLIP tags
perl /usr/local/CTK/bed2annotation.pl -dbkey hg19 -ss -big -region -v -summary HepG2.RBFOX2.pool.R2.tag.uniq.annot.summary.txt HepG2.RBFOX2.pool.R2.tag.uniq.rgb.bed HepG2.RBFOX2.pool.R2.tag.uniq.annot.txt
Make sure the current genome (hg19) is specified. Check the summary file (HepG2.RBFOX2.pool.R2.tag.uniq.annot.summary.txt) for the percentage of tags mapped to CDS, 3'UTR, introns, etc.
Generate bedgraph for visualization in the genome browser
perl /usr/local/CTK/tag2profile.pl -v -ss -exact -of bedgraph -n ″Unique Tag Profile″ HepG2.RBFOX2.pool.R2.tag.noRNA.uniq.rgb.bed HepG2.RBFOX2.pool.R2.tag.uniq.bedgraph
The output bedgraph file can be loaded into a genome browser for visualization of tag number at each genomic position.
Peak calling
cd ~/test mkdir cluster cd cluster ln -s ../mapping/HepG2.RBFOX2.pool.R2.tag.uniq.rgb.bed
Mode 1: Peak calling with no statistical significance
perl /usr/local/CTK/tag2peak.pl -big -ss -v --valley-seeking --valley-depth 0.9 HepG2.RBFOX2.pool.R2.tag.uniq.rgb.bed HepG2.RBFOX2.pool.R2.tag.uniq.peak.bed --out-boundary HepG2.RBFOX2.pool.R2.tag.uniq.peak.boundary.bed --out-half-PH HepG2.RBFOX2.pool.R2.tag.uniq.peak.halfPH.bed
Note 1: To annotate peaks with overlapping genes and repeat masked sequences and get genomic breakdown:
perl /usr/local/CTK/bed2annotation.pl -dbkey hg19 -ss -big -region -v -summary HepG2.RBFOX2.pool.R2.tag.uniq.peak.annot.summary.txt HepG2.RBFOX2.pool.R2.tag.uniq.peak.bed HepG2.RBFOX2.pool.R2.tag.uniq.peak.annot.txt
The output file HepG2.RBFOX2.pool.R2.tag.uniq.peak.annot.txt has the detailed annotation for each peak, and HepG2.RBFOX2.pool.R2.tag.uniq.peak.annot.summary.txt has summary statistics.
Note 2: Another useful option --valley-depth specifies the depth of the valley relative to the peak (0.5-1, 0.9 by default). One also has the option to merge peaks close to each other by specifying the distance between the peaks (e.g., -gap 20).
Note 3: Besides the peak boundaries (HepG2.RBFOX2.pool.R2.tag.uniq.peak.bed), one can also output cluster boundaries (--out-boundary) and half peak boundaries (--out-half-PH ) associated with each peak.
In this mode, the script will not assess the statistical significance of the peak height. If one needs this, an alternative mode is as follows:
Mode 2: Peak calling with statistical significance
perl /usr/local/CTK/tag2peak.pl -big -ss -v --valley-seeking -p 0.05 --valley-depth 0.9 --multi-test --dbkey hg19 HepG2.RBFOX2.pool.R2.tag.uniq.rgb.bed HepG2.RBFOX2.pool.R2.tag.uniq.peak.sig.bed --out-boundary HepG2.RBFOX2.pool.R2.tag.uniq.peak.sig.boundary.bed --out-half-PH HepG2.RBFOX2.pool.R2.tag.uniq.peak.sig.halfPH.bed
Note 1: To get the number of significant peaks:
wc -l HepG2.RBFOX2.pool.R2.tag.noRNA.uniq.peak.sig.bed 39482 HepG2.RBFOX2.pool.R2.tag.uniq.peak.sig.bed
Note 2: To search for a known binding motif, one first defines the center of each peak (based on width at peak).
#extract from -500 to +500
awk '{print $1"\t"int(($2+$3)/2)-500"\t"int(($2+$3)/2)+500"\t"$4"\t"$5"\t"$6}' HepG2.RBFOX2.pool.R2.tag.uniq.peak.sig.bed > HepG2.RBFOX2.pool.R2.tag.uniq.peak.sig.center.ext1k.bed
This bed file can be used to extract sequences around peak (pay attention to the strand), and search for enrichment of specific motif (e.g., UGCAUG) relative to the peak.
Note 3: One might want to count the number of tags overlapping with each cluster/peak for each sample (e.g., to evaluate correlation between replicates).
for f in HepG2.RBFOX2.rep1.R2 HepG2.RBFOX2.rep2.R2; do
perl /usr/local/CTK/tag2profile.pl -ss -region HepG2.RBFOX2.pool.R2.tag.uniq.peak.sig.boundary.bed -of bed -v $f.tag.uniq.bed $f.tag.uniq.peak.sig.boundary.count.bed
done
Download output files for peak calling here (25 MB).
CIMS
cd ~/test mkdir CIMS cd CIMS ln -s ../mapping/HepG2.RBFOX2.pool.R2.tag.uniq.rgb.bed ln -s ../mapping/HepG2.RBFOX2.pool.R2.tag.uniq.mutation.txt
Get specific types of mutations
Get specific types of mutations, such as deletions, substitutions, and insertions in unique CLIP tags
perl ~/czlab_src/CTK/getMutationType.pl -t del HepG2.RBFOX2.pool.R2.tag.uniq.mutation.txt HepG2.RBFOX2.pool.R2.tag.uniq.del.bed #deletions perl ~/czlab_src/CTK/getMutationType.pl -t ins HepG2.RBFOX2.pool.R2.tag.uniq.mutation.txt HepG2.RBFOX2.pool.R2.tag.uniq.ins.bed #insertions perl ~/czlab_src/CTK/getMutationType.pl -t sub --summary HepG2.RBFOX2.pool.R2.tag.uniq.mutation.stat.txt HepG2.RBFOX2.pool.R2.tag.uniq.mutation.txt HepG2.RBFOX2.pool.R2.tag.uniq.sub.bed #substitutions, as well as summary statistics of different types of mutations
It is always a good practice to look at the number of each type of mutation to see, e.g., compare the relative abundance of deletions to insertions. This information is provided in the summary statistics file when one specify the option --summary.
Get CIMS
Here, we use deletions as an example.
perl /usr/local/CTK/CIMS.pl -big -n 10 -p -outp HepG2.RBFOX2.pool.R2.tag.uniq.del.pos.stat.txt -v HepG2.RBFOX2.pool.R2.tag.uniq.rgb.bed HepG2.RBFOX2.pool.R2.tag.uniq.del.bed HepG2.RBFOX2.pool.R2.tag.uniq.del.CIMS.txt
By default, CIMS.pl will output all sites including those that are not statistically significant. This is recommended because one can play with stringency later.
Extract sites that are statistically significant.
awk '{if($9<=0.001) {print $0}}' HepG2.RBFOX2.pool.R2.tag.uniq.del.CIMS.txt | sort -k 9,9n -k 8,8nr -k 7,7n > HepG2.RBFOX2.pool.R2.tag.uniq.del.CIMS.s30.txt
cut -f 1-6 HepG2.RBFOX2.pool.R2.tag.uniq.del.CIMS.s30.txt > HepG2.RBFOX2.pool.R2.tag.uniq.del.CIMS.s30.bed
wc -l HepG2.RBFOX2.pool.R2.tag.uniq.del.CIMS.s30.bed 1403 HepG2.RBFOX2.pool.R2.tag.uniq.del.CIMS.s30.bed
This will keep only those with FDR<0.001. Significant sites will also be sorted by FDR, then by the number of tags with mutations, and then by the total number of overlapping tags. One might try different thresholds to get a balance of sensitivity/specificity (e.g. as judged from motif enrichment).
Note 1: Another parameter that might be useful to improve signal to noise is m/k (i.g., $8/$7 in awk)
Note 2: By default, the command line above will analyze mutations of size 1. Sometimes deletion of multiple nucleotides occurs and those will be ignored here. One can analyze mutations of size 2 by specifying -w 2. Substitutions are always reported as a single nucleotide (even when consecutive nucleotides are substituted), and insertions occur technically in one position and are thus treated as size 1.
Note 3: The number of significant CIMS might be slightly different when unique tags are provided in different orders (this could occur easily when multiple replicates are concatenated together). This is because the FDR estimation is based on permutation, which can change slightly depending on the order of tags.
Note 4: To examine enrichment of motif around CIMS:
awk '{print $1"\t"$2-10"\t"$3+10"\t"$4"\t"$5"\t"$6}' HepG2.RBFOX2.pool.R2.tag.uniq.del.CIMS.s30.bed > HepG2.RBFOX2.pool.R2.tag.uniq.del.CIMS.s30.21nt.bed
#+/-10 around CIMS
This BED file could be used to extract sequences around CIMS (pay attention to the strand), and search for enrichment of specific motif (e.g., UGCAUG for the case of RBFOX2) relative to the CIMS.
Download CIMS output files here (1.5 MB).
One can repeat these steps for the other types of mutations (i.e. substitutions and insertions). This is particularly relevant when one works with a new RBP, and it is unknown which type of mutations will be caused by cross linking.
CITS
cd ~/test mkdir CITS cd CITS ln -s ../mapping/HepG2.RBFOX2.pool.R2.tag.uniq.rgb.bed ln -s ../CIMS/HepG2.RBFOX2.pool.R2.tag.uniq.del.bed
perl ~/czlab_src/CTK/CITS.pl -big -p 0.001 --gap 25 -v HepG2.RBFOX2.pool.R2.tag.uniq.bed HepG2.RBFOX2.pool.R2.tag.uniq.del.bed HepG2.RBFOX2.pool.R2.tag.uniq.clean.CITS.s30.bed
In this script, we will look for reproducible CLIP tag start positions with more supporting tags than one would expect, which likely indicates crosslink induced truncation sites. For RBFOX2, since the vast majority of deletions are introduced because of cross linking, tags with deletions are treated as read-through tags and removed for CITS analysis.
Note 1: One can now perform motif enrichment analysis as described above in the CIMS section.
Note 2: In this particular example, we decided not to do Bonforroni correction, as we found this is too conservative. If necessary, this can be done by using option --multi-test.
Note 3: In the command line above, we opt to merge sites very close to each other and keep the most significant ones because CITS analysis tends to have more background when read through is frequent.
A few sites that span multiple nucleotides can be removed:
awk '{if($3-$2==1) {print $0}}' HepG2.RBFOX2.pool.R2.tag.uniq.clean.CITS.s30.bed > HepG2.RBFOX2.pool.R2.tag.uniq.clean.CITS.s30.singleton.bed
wc -l HepG2.RBFOX2.pool.R2.tag.uniq.clean.CITS.s30.bed HepG2.RBFOX2.pool.R2.tag.uniq.clean.CITS.s30.singleton.bed 14910 HepG2.RBFOX2.pool.R2.tag.uniq.clean.CITS.s30.bed 14823 HepG2.RBFOX2.pool.R2.tag.uniq.clean.CITS.s30.singleton.bed
Note 4: To examine enrichment of motif around CIMS:
awk '{print $1"\t"$2-10"\t"$3+10"\t"$4"\t"$5"\t"$6}' HepG2.RBFOX2.pool.R2.tag.uniq.clean.CITS.s30.singleton.bed > HepG2.RBFOX2.pool.R2.tag.uniq.clean.CITS.s30.singleton.21nt.bed
#+/-10 around CITS
This BED file could be used to extract sequences around CITS (pay attention to the strand), and search for enrichment of specific motif (e.g., UGCAUG for the case of Rbfox1-3) relative to the CITS.
Download CITS output files here (587 KB).
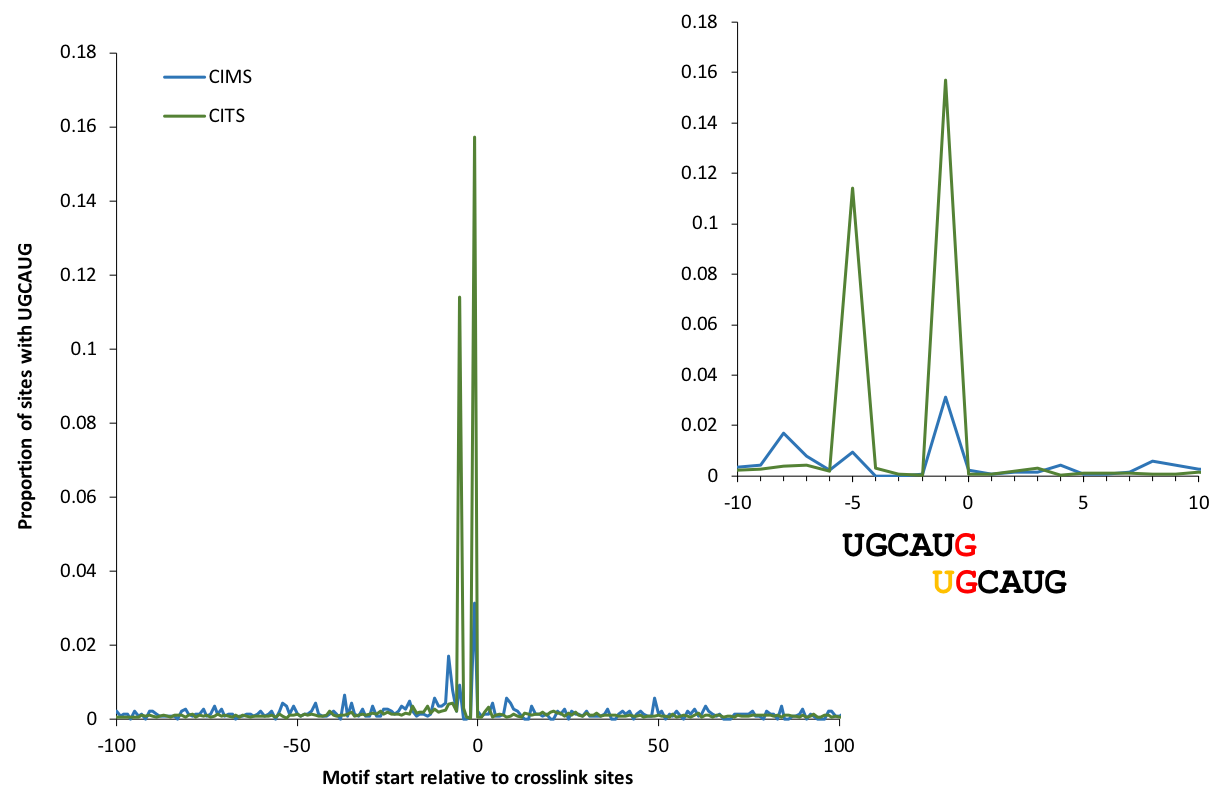
Motif enrichment
The enrichment of motif sites (when the motif is known) around CIMS or CITS provides a quantitative measure of the signal to noise ratio. The plot below shows the proportion of sites with the RBFOX2 binding UGCAUG motif starting at each position relative to CIMS/CITS (using tools not included in CTK). This analysis identifies G2 and G5 as the major crosslink sites between RBFOX2 and RNA.