Standard/BrdU-CLIP data analysis using CTK
From Zhang Laboratory
Contents
Introduction
This tutorial covers analysis of CLIP data generated by the standard CLIP protocol (Moore et al. 2014 Nat Protocols 9:263-293) and the BrdU-CLIP protocol (Weyn-Vanhentenryck et al. 2014 Cell Rep 6:1139-1152).
Reads from the standard CLIP protocol begin with a 5nt random barcode, followed by the actual cDNA tags of variable lengths, and possibly 3' adaptor sequences:
NNNNN[CLIP tag]GTGTCAGTCACTTCCAGCGGTCGTATGCCGTCTTCTGCTTG
where N5 is the degenerate (random) barcode, which is introduced before PCR amplification to serve as a unique molecular identifier (UMI).
With the BrdU-CLIP protocol, several CLIP libraries with different indexes are typically pooled together to be sequenced in one lane.
Reads from the BrdU-CLIP protocol begin with a 14 nucleotides that are sample index and random barcode (4 nt sample index+ a fixed base (either G or C)+ an 8-nt random barcode + a G), followed by the actual cDNA tags of variable lengths, and possibly 3' adaptor sequences.
SSSSSNNNNNNNNN[CLIP tag]AGCATACGGCAGAAGACGAAC
where S5 is the sample index and N9 is the degenerate barcode.
Sample dataset
Raw FASTQ files
In this documentation, we will use the sample mouse brain Rbfox1-3 CLIP data (Weyn-Vanhentenryck et al. Cell Reports 2014) as a guide. Two protocols (standard CLIP and BrdU-CLIP) were used to generate this dataset.
We will use ~/test as our working directory, and download fastq files under ~/test/fastq
mkdir ~/test; cd ~/test mkdir fastq; cd fastq
The raw sequence files from Illumina sequencing can be downloaded from SRA: http://www.ncbi.nlm.nih.gov/sra/?term=SRP035321.
We assume FASTQ files have been generated using the toolkit and renamed as follows: Fox1_1.fastq.gz, Fox1_2.fastq.gz, Fox1_3.fastq.gz, Fox2_1.fastq.gz, Fox2_2.fastq.gz, Fox2_3.fastq.gz, Fox2_4.fastq.gz, Fox3_1.fastq.gz, Fox3_2.fastq.gz, Fox3_3.fastq.gz, Fox3_4.fastq.gz, Fox3_5.fastq.gz under the fastq folder.
Fox1_1.fastq.gz, Fox2_1.fastq.gz, and Fox3_1.fastq.gz were generated from the Standard CLIP protocol, and the rest were generated from the BrdU CLIP protocol.
Output files of major steps generated in this tutorial
- Fully preprocessed FASTQ files (1.9 GB) : output files generated for use right before mapping.
- Unique CLIP tag and mutation files (506 MB) : output files generated after mapping and collapsing PCR duplicates.
- Peak calling files (26 MB) : output files after peak calling.
- CIMS output files (1.6 MB) : output files after CIMS analysis.
- CITS output files (641 KB) : output files after CITS analysis.
The number of reads in each file, after each step of processing, is summarized in Table 1 below.
| Protein | Sample | CLIP protocol | # of raw reads | # of filtered reads | # of trimmed reads | # of collapsed reads | # of mapped reads | # of unique tags |
|---|---|---|---|---|---|---|---|---|
| Fox1 | Fox1_1 | Standard | 128,502,865 | 124,979,444 | 113,958,244 | 4,953,805 | 3,713,799 | 357,094 |
| Fox1 | Fox1_2 | BrdU | 42,373,740 | 39,121,986 | 35,683,047 | 1,465,789 | 1,098,974 | 94,757 |
| Fox1 | Fox1_3 | BrdU | 36,103,020 | 32,500,181 | 29,682,907 | 2,112,635 | 1,468,725 | 161,487 |
| Fox1 | Fox1_4 | BrdU | 64,528,510 | 52,712,783 | 48,292,964 | 7,025,945 | 5,262,144 | 1,194,617 |
| Fox2 | Fox2_1 | Standard | 135,171,343 | 131,229,217 | 121,238,246 | 5,406,496 | 3,716,401 | 295,180 |
| Fox2 | Fox2_2 | BrdU | 42,744,272 | 39,351,367 | 35,005,375 | 1,314,878 | 968,115 | 83,358 |
| Fox2 | Fox2_3 | BrdU | 31,512,102 | 28,530,199 | 25,877,412 | 2,356,210 | 1,581,335 | 337,811 |
| Fox2 | Fox2_4 | BrdU | 46,383,375 | 40,317,796 | 29,418,988 | 2,761,500 | 1,803,408 | 165,645 |
| Fox3 | Fox3_1 | Standard | 181,578,804 | 173,884,893 | 162,979,630 | 10,405,177 | 7,742,410 | 877,256 |
| Fox3 | Fox3_2 | BrdU | 38,828,880 | 35,983,470 | 27,171,512 | 1,185,180 | 857,925 | 143,522 |
| Fox3 | Fox3_3 | BrdU | 31,774,690 | 29,095,065 | 26,332,663 | 2,295,078 | 1,669,958 | 399,878 |
| Fox3 | Fox3_4 | BrdU | 32,658,459 | 29,720,851 | 21,737,823 | 2,017,979 | 1,479,115 | 413,847 |
| Fox3 | Fox3_5 | BrdU | 58,175,879 | 47,324,888 | 42,941,116 | 6,200,102 | 4,551,275 | 1,076,782 |
| Total | 870,706,469 | 5,601,234 |
Read preprocessing
In all preprocessing steps prior to read alignment, operates on FASTQ files to take advantage of sequence quality scores. This is different from our previous software package CIMS.
Demultiplexing samples
The files downloaded from SRA have already be demultiplexed. Therefore the next step has already been performed. It is included for illustration purposes for the user who would like to start from a FASTQ file consisting of multiple libraries.
cd ~/test/fastq
Assuming that we have six samples with indexes GTCA, GCAT, ACTG, AGCT, GCAT, TCGA, the following script will extract reads for each library using a simple loop.
#!/bin/bash
#For illustration
index=(GTCA GCAT ACTG AGCT GCAT TCGA);
samples=(sample1 sample2 sample3 sample4 sample5 sample6);
for ((i=0;i<=5;i=i+1)); do
idx=${index[$i]}
name=${samples[$i]}
perl /usr/local/CTK/fastq_filter.pl -v -if sanger -index 0:$idx -of fastq raw.fastq.gz - | gzip -c > $name.fastq.gz
done
Alternatively, we can parallelize the process by submitting an array of jobs to SGE.
#!/bin/bash
#$ -t 1-6 -m a -cwd -N CLIP
#For illustration
index=(GTCA GCAT ACTG AGCT GCAT TCGA);
samples=(sample1 sample2 sample3 sample4 sample5 sample6);
idx=${index[$SGE_TASK_ID-1]}
name=${samples[$SGE_TASK_ID-1]}
perl /usr/local/CTK/fastq_filter.pl -v -if sanger -index 0:$idx -of fastq raw.fastq.gz - | gzip -c > $name.fastq.gz
The script /usr/local/CTK/fastq_filter.pl will extract reads with indicated index sequence (e.g., GTCA) starting from position 0 (specified by the -index option) in the read.
- Usage and additional explanation of fastq_filter.pl
Note 1: The reason we used the first four positions of the index for demultiplexing is due to historical confusion about the identity of the index sequences. It really should be 5nt, but it does not make a substantial difference.
Note 2: This script can also be used to demultiplex samples from other CLIP protocols as long as the position of the sample index relative to the read start is fixed.
Note 3: To get usage information, just run the script without any parameters (the same for the other scripts in the package).
Read quality filtering
We filter out low quality reads because those reads can introduce mapping errors and background. This will typically inflate the number of unique tags after removal of PCR duplicates.
cd ~/test mkdir filtering cd filtering
The same fastq_filter.pl script will extract reads passing quality filters for each library (of standard CLIP-seq data) using a simple loop.
#!/bin/bash for f in Fox1_1 Fox2_1 Fox3_1 do perl /usr/local/CTK/fastq_filter.pl -v -if sanger -f mean:0-29:20 -of fastq ../fastq/$f.fastq.gz - | gzip -c > $f.fastq.gz done
This is the same thing, but using a Sun Grid Engine to execute this UNIX job on a remote machine.
#!/bin/bash
#$ -t 1-3 -m a -cwd -N CLIP
files=(Fox1_1 Fox2_1 Fox3_1);
f=${files[$SGE_TASK_ID-1]}
perl /usr/local/CTK/fastq_filter.pl -v -if sanger -f mean:0-29:20 -of fastq ../fastq/$f.fastq.gz - | gzip -c > $f.fastq.gz
This script filters raw reads based on quality scores. -f mean:0-29-20 specifies a mean score of 20 or above in the first 30 bases (i.e., positions 0-29 for Standard CLIP), which includes 5 positions with sample indexes and the random barcode, followed by 25 positions with the actual CLIP tag (and 3' adaptors is the CLIP tag is shorter than 25 nt).
The filtering should be run again on the BrdU CLIP (or iCLIP) samples slightly differently.
#!/bin/bash
#$ -t 1-10 -m a -cwd -N CLIP
files=(Fox1_2 Fox1_3 Fox1_4 Fox2_2 Fox2_3 Fox2_4 Fox3_2 Fox3_3 Fox3_4 Fox3_5);
f=${files[$SGE_TASK_ID-1]}
perl /usr/local/CTK/fastq_filter.pl -v -if sanger -f mean:0-38:20 -of fastq ../fastq/$f.fastq.gz - | gzip -c > $f.fastq.gz
-f mean:0-38-20 specifies a mean score of 20 or above in the first 39 bases (i.e., positions 0-38 for BrdU CLIP), which includes 14 positions with sample indexes and the random barcode, followed by 25 positions with the actual CLIP tag.
Note 1: As a good practice, always check the number of reads in the raw files and compare the number of reads left after filtering to confirm if the numbers are what you would expect. This can be done by running the command:
#e.g., for raw reads
for f in `ls ../fastq/*.fastq.gz`; do
c=`zcat $f | wc -l`
c=$((c/4))
echo $f $c
done
The wc command will give the number of lines (divide by 4 to get the actual read number). You can compare the numbers you obtain with the numbers we provided in the table above to ensure you are getting the same results.
Note 2: Demultiplexing and quality filtering can be combined into a single step by specifying -index and -f at the same time.
perl /usr/local/CTK/fastq_filter.pl -v -if sanger -index 0:GTCA -f mean:0-38:20 -of fastq ../fastq/in.fastq.gz - | gzip -c > out.fastq.gz
Trimming of 3' adapter sequences
For long reads that are common now, collapsing before trimming is not very helpful. Therefore, we trim the 3' adaptor first (which can vary depending on the specific CLIP protocol performed).
For each sample, run the following command using tools provided by the Fastx Toolkit.
#!/bin/bash
#$ -t 1-3 -m a -cwd -N CLIP
files=(Fox1_1 Fox2_1 Fox3_1);
f=${files[$SGE_TASK_ID-1]}
zcat $f.fastq.gz | fastx_clipper -a GTGTCAGTCACTTCCAGCGG -l 20 -n | fastq_quality_trimmer -t 5 -l 20 -z -o $f.trim.fastq.gz
#note that the 3' adaptor will vary for other CLIP protocol variations.
This command will remove the 3' adaptor and also remove extremely low quality bases (score < 5). We require 20 nt (5 nt for random barcode and 15 nt for CLIP tags) after trimming.
Here one can consider including the whole 3' adapter sequence (including the part introduced by PCR; GTGTCAGTCACTTCCAGCGGTCGTATGCCGTCTTCTGCTTG) for trimming.
You can do the same for the BrdU CLIP files:
#!/bin/bash
#$ -t 1-10 -m a -cwd -N CLIP
files=(Fox1_2 Fox1_3 Fox1_4 Fox2_2 Fox2_3 Fox2_4 Fox3_2 Fox3_3 Fox3_4 Fox3_5);
f=${files[$SGE_TASK_ID-1]}
zcat $f.fastq.gz | fastx_clipper -a TCGTATGCCGTCTTCTGCTTG -l 29 -n | fastq_quality_trimmer -t 5 -l 29 -z -o $f.trim.fastq.gz
We require 29 nt (14 nt for sample index+random barcode and 15 nt for CLIP tags) after trimming.
For fastx_clipper usage information:
fastx_clipper -h
Note 1: It is good to check the number of reads by running the command:
for f in `ls *.trim.fastq.gz`; do
c=`zcat $f | wc -l`
c=$((c/4))
echo $f $c
done
Note 2: we may use cutadapt for adapter trimming in the future.
cutadapt --times 1 -e 0.1 -O 1 --quality-cutoff 5 -m 20 -a GTGTCAGTCACTTCCAGCGG -o $f.trim.fastq.gz ../fastq/$f.fastq.gz > $f.cutadpt.log #for standard CLIP as an example
Collapse exact duplicates
If multiple reads have exactly the same sequence, only one is kept.
#!/bin/bash
#$ -t 1-13 -m a -cwd -N CLIP
files=(Fox1_1 Fox1_2 Fox1_3 Fox1_4 Fox2_1 Fox2_2 Fox2_3 Fox2_4 Fox3_1 Fox3_2 Fox3_3 Fox3_4 Fox3_5);
f=${files[$SGE_TASK_ID-1]}
perl /usr/local/CTK/fastq2collapse.pl $f.trim.fastq.gz - | gzip -c > $f.trim.c.fastq.gz
- Usage and additional explanation of fastq2collapse.pl.
Note 1: It is good to check the number of reads by running the command:
for f in `ls *.trim.c.fastq.gz`; do
c=`zcat $f | wc -l`
c=$((c/4))
echo $f $c
done
Strip random barcode (UMI)
The following script removes the 5' degenerate barcode from the sequence and attach it to the sequence ID.
#The barcode for standard CLIP is 5 nucleotides long
for f in Fox1_1 Fox2_1 Fox3_1
do
perl /usr/local/CTK/stripBarcode.pl -format fastq -len 5 $f.trim.c.fastq.gz - | gzip -c > $f.trim.c.tag.fastq.gz
done
#the barcode for BrdU CLIP is 14 nucleotides long
for f in Fox1_2 Fox1_3 Fox1_4 Fox2_2 Fox2_3 Fox2_4 Fox3_2 Fox3_3 Fox3_4 Fox3_5
do
perl /usr/local/CTK/stripBarcode.pl -format fastq -len 14 $f.trim.c.fastq.gz - | gzip -c > $f.trim.c.tag.fastq.gz
done
Note 1: For BrdU CLIP (and other CLIP protocols with both sample index and random barcode that are next to each other), we treat sample index as part of the random barcode here.
- Usage and additional explanation of stripBarcode.pl.
After this step, one can get the distribution of tag length for diagnostic purposes.
zcat $f.trim.c.tag.fastq.gz | awk '{if(NR%4==2) {print length($0)}}' | sort -n | uniq -c | awk '{print $2"\t"$1}' > $f.trim.c.tag.seqlen.stat.txt
Download preprocessed FASTQ files here (1.9 GB)
Read mapping & parsing
Read mapping
We assume the reference index has been prepared and is available under /genomes/mm10/bwa/
cd ~/test mkdir mapping cd mapping
Run bwa to align the reads to the reference genome.
#!/bin/bash
#$ -t 1-13 -m a -cwd -N CLIP
files=(Fox1_1 Fox1_2 Fox1_3 Fox1_4 Fox2_1 Fox2_2 Fox2_3 Fox2_4 Fox3_1 Fox3_2 Fox3_3 Fox3_4 Fox3_5);
f=${files[$SGE_TASK_ID-1]}
bwa aln -t 4 -n 0.06 -q 20 /genomes/mm10/bwa/mm10.fa ../filtering/$f.trim.c.tag.fastq.gz > $f.sai
bwa samse /genomes/mm10/bwa/mm10.fa $f.sai ../filtering/$f.trim.c.tag.fastq.gz | gzip -c > $f.sam.gz
#Alternatively, the two steps can also be combined, using the following command:
bwa aln -t 4 -n 0.06 -q 20 /genomes/mm10/bwa/mm10.fa ../filtering/$f.trim.c.tag.fastq.gz | bwa samse /genomes/mm10/bwa/mm10.fa - ../filtering/$f.trim.c.tag.fastq.gz | gzip -c > $f.sam.gz
The option -n 0.06 specifies slightly more stringent criteria than the default. The number of allowed mismatches (substitutions or indels) depending on read length is as follows:
[bwa_aln] 17bp reads: max_diff = 1 [bwa_aln] 20bp reads: max_diff = 2 [bwa_aln] 45bp reads: max_diff = 3 [bwa_aln] 73bp reads: max_diff = 4 [bwa_aln] 104bp reads: max_diff = 5 [bwa_aln] 137bp reads: max_diff = 6 [bwa_aln] 172bp reads: max_diff = 7 [bwa_aln] 208bp reads: max_diff = 8 [bwa_aln] 244bp reads: max_diff = 9
The -q option is used to trim low quality reads (an average of 20 or below). However, this does not seem to do too much after the trimming steps above.
Parsing SAM file
#!/bin/bash
#$ -t 1-13 -m a -cwd -N CLIP
files=(Fox1_1 Fox1_2 Fox1_3 Fox1_4 Fox2_1 Fox2_2 Fox2_3 Fox2_4 Fox3_1 Fox3_2 Fox3_3 Fox3_4 Fox3_5);
f=${files[$SGE_TASK_ID-1]}
perl /usr/local/CTK/parseAlignment.pl -v --map-qual 1 --min-len 18 --mutation-file $f.mutation.txt $f.sam.gz $f.tag.bed
This will keep only unique mappings (with MAPQ >=1) and a minimal mapping size of 18 nt.
Note 1: In the tag bed file, the 5th column records the number of mismatches (substitutions) in each read
Note 2: Other aligners might not use a positive MAPQ as an indication of unique mapping.
Another useful option is --indel-to-end, which specifies the number of nucleotides towards the end from which indels should not be called (default=5 nt).
Note 3: The parsing script relies on MD tags, which is an optional field without strict definition in SAM file format specification. Some aligners might have slightly different format how they report mismatches. If other aligners than bwa is used, one should run the following command:
samtools view -bS $f.sam | samtools sort - $f.sorted samtools fillmd $f.sorted.bam /genomes/mm10/bwa/mm10.fa | gzip -c > $f.sorted.md.sam.gz
This will ensure the sam file gets parsed properly.
- Usage and additional explanation of parseAlignment.pl.
Note 4: Keep track what proportion of reads can be mapped uniquely.
wc -l *.tag.bed
Note 5: the format of the mutation file is essentially the same as before.
Gettiing unique CLIP tags by collapsing PCR duplicates
It is critical to collapse PCR duplicates, not only for the exact duplicates collapsed above, but also for those with slight differences due to sequencing errors. These sequencing errors can also occur in the random barcode, so a naive method to collapse the same barcode is frequently insufficient.
#!/bin/bash
#$ -t 1-13 -m a -cwd -N CLIP
files=(Fox1_1 Fox1_2 Fox1_3 Fox1_4 Fox2_1 Fox2_2 Fox2_3 Fox2_4 Fox3_1 Fox3_2 Fox3_3 Fox3_4 Fox3_5);
f=${files[$SGE_TASK_ID-1]}
perl /usr/local/CTK/tag2collapse.pl -v -big --random-barcode -EM 30 --seq-error-model alignment -weight --weight-in-name --keep-max-score --keep-tag-name $f.tag.bed $f.tag.uniq.bed
- Usage and additional explanation of tag2collapse.pl.
In the command above, tags mapped to the same genomic positions (i.e., the same start coordinates for the 5' end of RNA tag) will be collapsed together if they share the same or "similar" barcodes. A model-based algorithm is used to identify "sufficiently distinct" barcodes. This algorithm was described in detail in the following paper:
Darnell JC, et al. FMRP Stalls Ribosomal Translocation on mRNAs Linked to Synaptic Function and Autism. Cell. 2011; 146:247–261.
Compared to the original algorithm, the current implementation estimates sequencing error from aligned reads, which is then fixed during the iterative EM procedure.
Note 1: Sequencing errors in the degenerate barcodes are estimated from results of read alignment. The number of substitutions in each read must be provided in the 5th column.
Note 2: Note that the read ID in the input BED file (in the 4th column) must take the form READ#x#NNNNN, where x is the number of exact duplicates and NNNNN is the bar-code nucleotide sequence appended to read IDs in previous steps. Read IDs that are not in this format will generate an error.
Note 3: As a diagnostic step, get the length distribution of unique tags, which should be a more faithful representation of the library:
for f in Fox1_1 Fox1_2 Fox1_3 Fox1_4 Fox2_1 Fox2_2 Fox2_3 Fox2_4 Fox3_1 Fox3_2 Fox3_3 Fox3_4 Fox3_5
do
awk '{print $3-$2}' $f.tag.uniq.bed | sort -n | uniq -c | awk '{print $2"\t"$1}' > $f.uniq.len.dist.txt
done
Get the mutations in unique tags
python /usr/local/CTK/joinWrapper.py $f.mutation.txt $f.tag.uniq.bed 4 4 N $f.tag.uniq.mutation.txt #This script is from galaxy, and included for your convenience #The parameters 4 4 indicate the columns in the two input file used to join the two, and N indicates that only paired rows should be printed
- Usage and additional explanation of selectRow.pl.
Table 2 summarizes the number of unique mutations of different types in each sample.
| Protein | Sample | CLIP protocol | # of unique tags | Total # of mutations in unique tags | Deletions | Insertions | Substitutions |
|---|---|---|---|---|---|---|---|
| Fox1 | Fox1_1 | Standard | 357,094 | 148,096 | 30,679 | 4,705 | 112,812 |
| Fox1 | Fox1_2 | BrdU | 94,757 | 34,615 | 2,743 | 2,022 | 29,850 |
| Fox1 | Fox1_3 | BrdU | 161,487 | 57,879 | 7,352 | 3,138 | 47,389 |
| Fox1 | Fox1_4 | BrdU | 1,194,617 | 396,076 | 44,013 | 11,019 | 341,044 |
| Fox2 | Fox2_1 | Standard | 295,180 | 136,908 | 26,795 | 6,136 | 103,977 |
| Fox2 | Fox2_2 | BrdU | 83,358 | 28,167 | 1,192 | 1,832 | 25,143 |
| Fox2 | Fox2_3 | BrdU | 337,811 | 90,267 | 10,912 | 3,442 | 75,913 |
| Fox2 | Fox2_4 | BrdU | 165,645 | 74,782 | 2,293 | 5,010 | 67,479 |
| Fox3 | Fox3_1 | Standard | 877,256 | 337,181 | 51,541 | 13,896 | 271,744 |
| Fox3 | Fox3_2 | BrdU | 143,522 | 42,843 | 6,060 | 2,059 | 34,724 |
| Fox3 | Fox3_3 | BrdU | 399,878 | 94,415 | 17,008 | 3,500 | 73,907 |
| Fox3 | Fox3_4 | BrdU | 413,847 | 93,007 | 17,197 | 3,153 | 72,657 |
| Fox3 | Fox3_5 | BrdU | 1,076,782 | 420,822 | 38,790 | 10,798 | 371,234 |
| Total | 5,601,234 | 1,955,158 | 256,575 | 70,710 | 1,627,873 |
Merging biological replicates
After getting the unique tags of each library, one might concatenate biological replicates, which are distinguished by different colors. As an example:
perl /usr/local/CTK/bed2rgb.pl -v -col "128,0,0" Fox1_1.tag.uniq.bed Fox1_1.tag.uniq.rgb.bed #as one example #repeat the above step for all other uniq.bed files to generate rgb.bed files, but use different rgb colors "x,x,x" (see http://www.rapidtables.com/web/color/RGB_Color.htm or other charts) cat *tag.uniq.rgb.bed > Fox.pool.tag.uniq.rgb.bed cat *tag.uniq.mutation.txt > Fox.pool.tag.uniq.mutation.txt
- Usage and additional explanation of bed2rgb.pl.
We also concatenate the BrdU CLIP BED files separately for CITS analysis downstream in this pipeline.
cat Fox1_2.tag.uniq.rgb.bed Fox1_3.tag.uniq.rgb.bed Fox1_4.tag.uniq.rgb.bed Fox2_2.tag.uniq.rgb.bed Fox2_3.tag.uniq.rgb.bed Fox2_4.tag.uniq.rgb.bed Fox3_2.tag.uniq.rgb.bed Fox3_3.tag.uniq.rgb.bed Fox3_4.tag.uniq.rgb.bed Fox3_5.tag.uniq.rgb.bed > BrdU.Fox.pool.tag.uniq.rgb.bed cat Fox1_2.tag.uniq.mutation.txt Fox1_3.tag.uniq.mutation.txt Fox1_4.tag.uniq.mutation.txt Fox2_2.tag.uniq.mutation.txt Fox2_3.tag.uniq.mutation.txt Fox2_4.tag.uniq.mutation.txt Fox3_2.tag.uniq.mutation.txt Fox3_3.tag.uniq.mutation.txt Fox3_4.tag.uniq.mutation.txt Fox3_5.tag.uniq.mutation.txt > BrdU.Fox.pool.tag.uniq.mutation.txt wc -l BrdU.Fox.pool.tag.uniq.rgb.bed BrdU.Fox.pool.tag.uniq.mutation.txt 4071704 BrdU.Fox.pool.tag.uniq.rgb.bed 1332873 BrdU.Fox.pool.tag.uniq.mutation.txt
Download unique tag and mutation files here (506 MB).
Annotating and visualizing CLIP tags
Get genomic distribution of CLIP tags
perl /usr/local/CTK/bed2annotation.pl -dbkey mm10 -ss -big -region -v -summary Fox.pool.tag.uniq.annot.summary.txt Fox.pool.tag.uniq.rgb.bed Fox.pool.tag.uniq.annot.txt
Make sure the current genome (mm10) is specified.
Check the summary file (Fox.pool.tag.uniq.annot.summary.txt) for the percentage of tags mapped to CDS, 3'UTR, introns, etc.
- Usage and additional explanation of bed2annotation.pl.
Generate bedgraph for visualization in the genome browser
perl /usr/local/CTK/tag2profile.pl -v -big -ss -exact -of bedgraph -n ″Unique Tag Profile″ Fox.pool.tag.uniq.rgb.bed Fox.pool.tag.uniq.bedgraph
- Usage and additional explanation of tag2profile.pl.
The output bedgraph file can be loaded into a genome browser for visualization of tag number at each genomic position.
Peak calling
cd ~/test mkdir cluster cd cluster ln -s ../mapping/Fox.pool.tag.uniq.rgb.bed
CTK provides two modes of peak calling. The first mode is peak calling without statistical assessment, which will find all potential peaks. The second mode is to find peaks with peak height more than one expected by chance.
Mode 1: Peak calling without statistical assessment
perl /usr/local/CTK/tag2peak.pl -big -ss -v --valley-seeking --valley-depth 0.9 Fox.pool.tag.uniq.rgb.bed Fox.pool.tag.uniq.peak.bed --out-boundary Fox.pool.tag.uniq.peak.boundary.bed --out-half-PH Fox.pool.tag.uniq.peak.halfPH.bed
The command line above searches for all potential peaks using a "valley seeking" algorithm. A peak is called if valleys of certain depths are found on both sides so that it is separated from other peaks. The results are similar to those generated by the original clustering algorithm. However, this valley seeking algorithm is advantageous because it is able to separate neighboring peaks without zero-tag gaps.
Note 1: To annotate peaks with overlapping genes and repeat masked sequences and get genomic breakdown:
perl /usr/local/CTK/bed2annotation.pl -dbkey mm10 -ss -big -region -v -summary Fox.pool.tag.uniq.peak.annot.summary.txt Fox.pool.tag.uniq.peak.bed Fox.pool.tag.uniq.peak.annot.txt
The output file Fox.pool.tag.uniq.peak.annot.txt has the detailed annotation for each peak, and Fox.pool.tag.uniq.peak.annot.summary.txt has summary statistics.
Note 2: Another useful option --valley-depth specifies the depth of the valley relative to the peak (0.5-1, 0.9 by default). One also has the option to merge peaks close to each other by specifying the distance between the peaks (e.g., -gap 20).
Note 3: Besides the peak boundaries (Fox.pool.tag.uniq.peak.bed), one can also output cluster boundaries (--out-boundary) and half peak boundaries (--out-half-PH ) associated with each peak.
In this mode, the script will not assess the statistical significance of the peak height. If one needs this, an alternative mode is as follows:
Mode 2: Peak calling with statistical assessment
perl /usr/local/CTK/tag2peak.pl -big -ss -v --valley-seeking -p 0.05 --valley-depth 0.9 --dbkey mm10 --multi-test Fox.pool.tag.uniq.rgb.bed Fox.pool.tag.uniq.peak.sig.bed --out-boundary Fox.pool.tag.uniq.peak.sig.boundary.bed --out-half-PH Fox.pool.tag.uniq.peak.sig.halfPH.bed
Here, the script searches for significant peaks by considering one specific genic region as a unit for permutations to estimate expected peak height (scan statistics is used here, so the permutation is only conceptual).
It is important to note that in this mode, only clusters overlapping with annotated genes are kept.
In both cases, the three output files provide the positions of peak (many times it is 1nt, but not always), the peak width at half peak height, and the boundaries of each peak. In practice, it appears that the center of peak or half-peak provides the most robust/precise indicator of the binding sites (although a shift might be present because of CITS, which can be adjusted after one examines the enrichment of the motif).
Note that the score in column 5 of this uniq.peak.sig.bed file is peak height (PH - see column 4 - and PH0 is the expected PH/background).
Note 1: To get the number of significant peaks:
wc -l Fox.pool.tag.uniq.peak.sig.bed 29076 Fox.pool.tag.uniq.peak.sig.bed
Note 2: To search for a known binding motif, one first defines the center of each peak (based on width at half PH).
awk '{print $1"\t"int(($2+$3)/2)-500"\t"int(($2+$3)/2)+500"\t"$4"\t"$5"\t"$6}' Fox.pool.tag.uniq.peak.sig.halfPH.bed > Fox.pool.tag.uniq.peak.sig.halfPH.center.ext1k.bed
#+/-500 around halfPH center
This bed file can be used to extract sequences around peak (pay attention to the strand), and search for enrichment of specific motif (e.g., UGCAUG) relative to the peak.
Note we locate the peak center position from the halfPH coordinates. An alternative is to use the center of the peak position defined in Fox.pool.tag.uniq.peak.sig.bed.
Note 3: One might want to count the number of tags overlapping with each cluster/peak for each sample (e.g., to evaluate correlation between replicates).
perl /usr/local/CTK/tag2profile.pl -ss -region Fox.pool.tag.uniq.peak.sig.boundary.bed -of bed -v $f.tag.uniq.bed $f.tag.uniq.peak.sig.boundary.count.bed
Download output files for peak calling here (26 MB).
CIMS analysis
cd ~/test mkdir CIMS cd CIMS ln -s ../mapping/Fox.pool.tag.uniq.rgb.bed ln -s ../mapping/Fox.pool.tag.uniq.mutation.txt
Get specific types of mutations
Get specific types of mutations, such as deletions, substitutions, and insertions in unique CLIP tags
perl /usr/local/CTK/getMutationType.pl -t del Fox.pool.tag.uniq.mutation.txt Fox.pool.tag.uniq.del.bed #deletions perl /usr/local/CTK/getMutationType.pl -t ins Fox.pool.tag.uniq.mutation.txt Fox.pool.tag.uniq.ins.bed #insertions perl /usr/local/CTK/getMutationType.pl -t sub --summary Fox.pool.tag.uniq.mutation.stat.txt Fox.pool.tag.uniq.mutation.txt Fox.pool.tag.uniq.sub.bed #substitutions, as well as summary statistics of different types of mutations
It is always a good practice to look at the number of each type of mutation to see, e.g., compare the relative abundance of deletions to insertions. This information is provided in the summary statistics file when one specify the option --summary.
Get CIMS
Here we use deletions as an example.
perl /usr/local/CTK/CIMS.pl -big -n 10 -p -outp Fox.pool.tag.uniq.del.pos.stat.txt -v -c cache_del Fox.pool.tag.uniq.rgb.bed Fox.pool.tag.uniq.del.bed Fox.pool.tag.uniq.del.CIMS.txt
By default, CIMS.pl will output all sites including those that are not statistically significant. This is recommended because one can play with stringency later.
- Usage and additional explanation of CIMS.pl.
awk '{if($9<=0.001) {print $0}}' Fox.pool.tag.uniq.del.CIMS.txt | sort -k 9,9n -k 8,8nr -k 7,7n > Fox.pool.tag.uniq.del.CIMS.s30.txt
cut -f 1-6 Fox.pool.tag.uniq.del.CIMS.s30.txt > Fox.pool.tag.uniq.del.CIMS.s30.bed
wc -l Fox.pool.tag.uniq.del.CIMS.s30.bed 5707 Fox.pool.tag.uniq.del.CIMS.s30.bed
This will keep only those with FDR<0.001. Significant sites will also be sorted by FDR, then by the number of tags with mutations, and then by the total number of overlapping tags. One might try different thresholds to get a balance of sensitivity/specificity (e.g. as judged from motif enrichment).
Note 1: Another parameter that might be useful to improve signal to noise is m/k (i.g., $8/$7 in awk)
Note 2: By default, the command line above will analyze mutations of size 1. Sometimes deletion of multiple nucleotides occurs and those will be ignored here. One can analyze mutations of size 2 by specifying -w 2. Substitutions are always reported as a single nucleotide (even when consecutive nucleotides are substituted), and insertions occur technically in one position and are thus treated as size 1.
Note 3: The number of significant CIMS might be slightly different when unique tags are provided in different orders (this could occur easily when multiple replicates are concatenated together). This is because the FDR estimation is based on permutation, which can change slightly depending on the order of tags.
Note 4: To examine enrichment of motif around CIMS:
awk '{print $1"\t"$2-10"\t"$3+10"\t"$4"\t"$5"\t"$6}' Fox.pool.tag.uniq.del.CIMS.s30.bed > Fox.pool.tag.uniq.del.CIMS.s30.21nt.bed
#+/-10 around CIMS
This BED file could be used to extract sequences around CIMS (pay attention to the strand), and search for enrichment of specific motif (e.g., UGCAUG for the case of Rbfox1-3) relative to the CIMS.
Download CIMS output files here (1.6 MB).
One can repeat these steps for the other types of mutations (i.e. substitutions and insertions). This is particularly relevant when one works with a new RBP, and it is unknown which type of mutations will be caused by cross linking.
CITS analysis
Only certain variations of the CLIP protocol allow you to perform CITS analysis (e.g. iCLIP, BrdU CLIP, etc).
Therefore, link only the BrdU CLIP files (i.e. exclude the Standard CLIP: Fox1_1, Fox2_1, Fox3_1).
cd ~/test mkdir CITS cd CITS ln -s ../mapping/BrdU.Fox.pool.tag.uniq.rgb.bed ./ ln -s ../CIMS/BrdU.Fox.pool.tag.uniq.del.bed ./
perl /usr/local/CTK/CITS.pl -big -p 0.001 --gap 25 -v BrdU.Fox.pool.tag.uniq.bed BrdU.Fox.pool.tag.uniq.del.bed BrdU.Fox.pool.tag.uniq.CITS.s30.bed
In this script, we will look for reproducible CLIP tag start positions with more supporting tags than one would expect, which likely indicates crosslink induced truncation sites. For Rbfox, since the vast majority of deletions are introduced because of cross linking, tags with deletions are treated as read-through tags and removed for CITS analysis.
Note 1: One can now perform motif enrichment analysis as described above in the CIMS section.
Note 2: In this particular example, we decided not to do Bonforroni correction, as we found this is too conservative. If necessary, this can be done by using option --multi-test.
Note 3: In the command line above, we opt to merge sites very close to each other and keep the most significant ones because CITS analysis tends to have more background when read through is frequent.
A few sites that span multiple nucleotides can be removed:
awk '{if($3-$2==1) {print $0}}' BrdU.Fox.pool.tag.uniq.CITS.s30.bed > BrdU.Fox.pool.tag.uniq.CITS.s30.singleton.bed
wc -l BrdU.Fox.pool.tag.uniq.CITS.s30.bed BrdU.Fox.pool.tag.uniq.CITS.s30.singleton.bed 16669 BrdU.Fox.pool.tag.uniq.CITS.s30.bed 16627 BrdU.Fox.pool.tag.uniq.CITS.s30.singleton.bed
Note 4: To examine enrichment of motif around CIMS:
awk '{print $1"\t"$2-10"\t"$3+10"\t"$4"\t"$5"\t"$6}' BrdU.Fox.pool.tag.uniq.CITS.s30.singleton.bed > BrdU.Fox.pool.tag.uniq.CITS.s30.singleton.21nt.bed
#+/-10 around CITS
This BED file could be used to extract sequences around CITS (pay attention to the strand), and search for enrichment of specific motif (e.g., UGCAUG for the case of Rbfox1-3) relative to the CITS.
Download CITS output files here (641 KB).
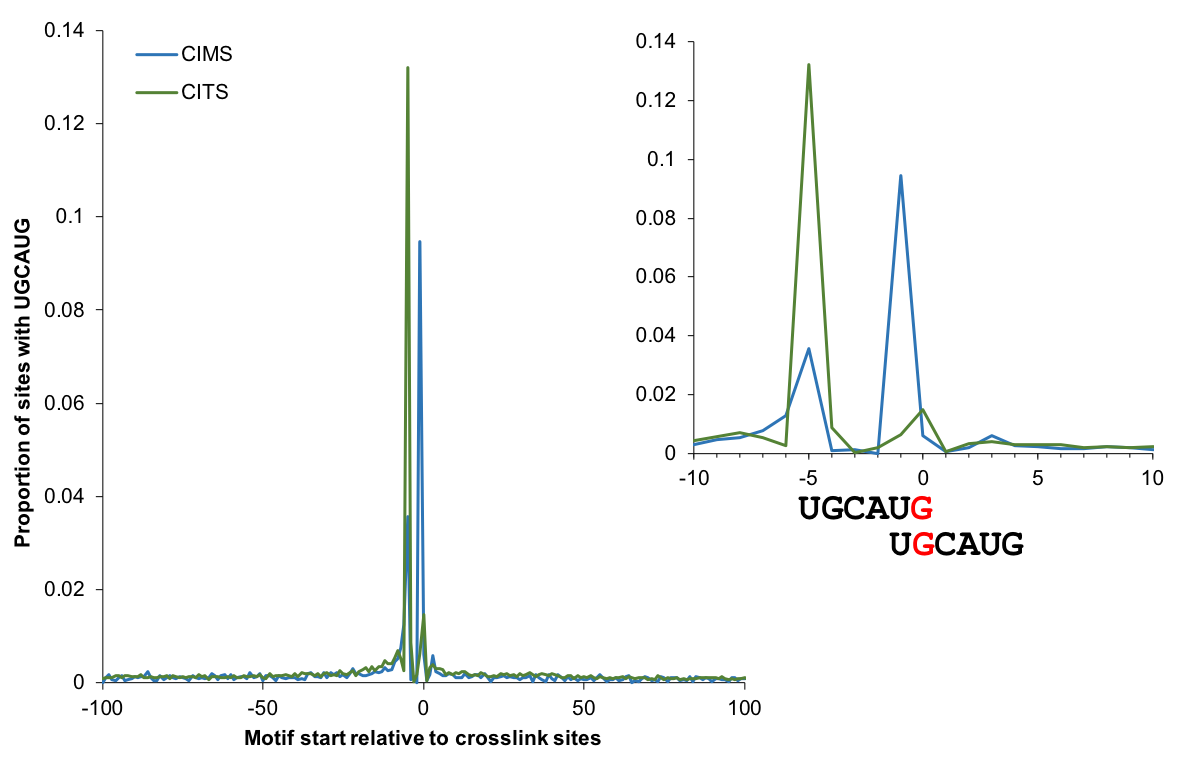
Motif enrichment
The enrichment of motif sites (when the motif is known) around CIMS or CITS provides a quantitative measure of the signal to noise ratio. The plot below shows the proportion of sites with the Rbfox binding UGCAUG motif starting at each position relative to CIMS/CITS (using tools not included in CTK). This analysis identifies G2 and G5 as the major crosslink sites between Rbfox and RNA.