Research
From Zhang Laboratory
We are working on RNA at the interface of Systems Biology, Data Science and Molecular Neuroscience. Our research has been funded by various government and private funding agencies.
![]()
![]()
![]()
![]()
![]()
![]()
![]()
Overview
|
Post-transcriptional regulation at the RNA level has profound impact on gene expression, especial for the development and function of the nervous system. Such regulation is dictated by interaction of at least several hundred RNA-binding proteins (RBPs) with their target transcripts, or RNA-regulatory networks. Our current work focus on neuron-specific alternative splicing, one critical step of RNA regulation. Our research interests range from basic understanding of the specificity of protein-RNA interaction, organization of splicing regulatory networks, function of specific splice variants, impact of genetic variants or mutation on splicing regulation, and RNA-based precision medicine. To approach the research goals, our lab regularly uses a variety of experimental and computational approaches and techniques, including CRISPR-based genome engineering, high-throughput screening, deep sequencing, probabilistic modeling and machine learning (e.g., Bayesian networks and deep learning). The lab uses both cell-based (e.g., mouse ESCs and human iPSCs and directed neuronal differentiation) and mouse models. Work in his lab has been funded by multiple Institutes at NIH, Simons Foundation, and Columbia Precision Medicine Initiative.
|
 |
RNA-based precision medicine
|
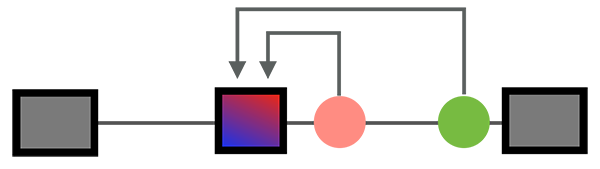
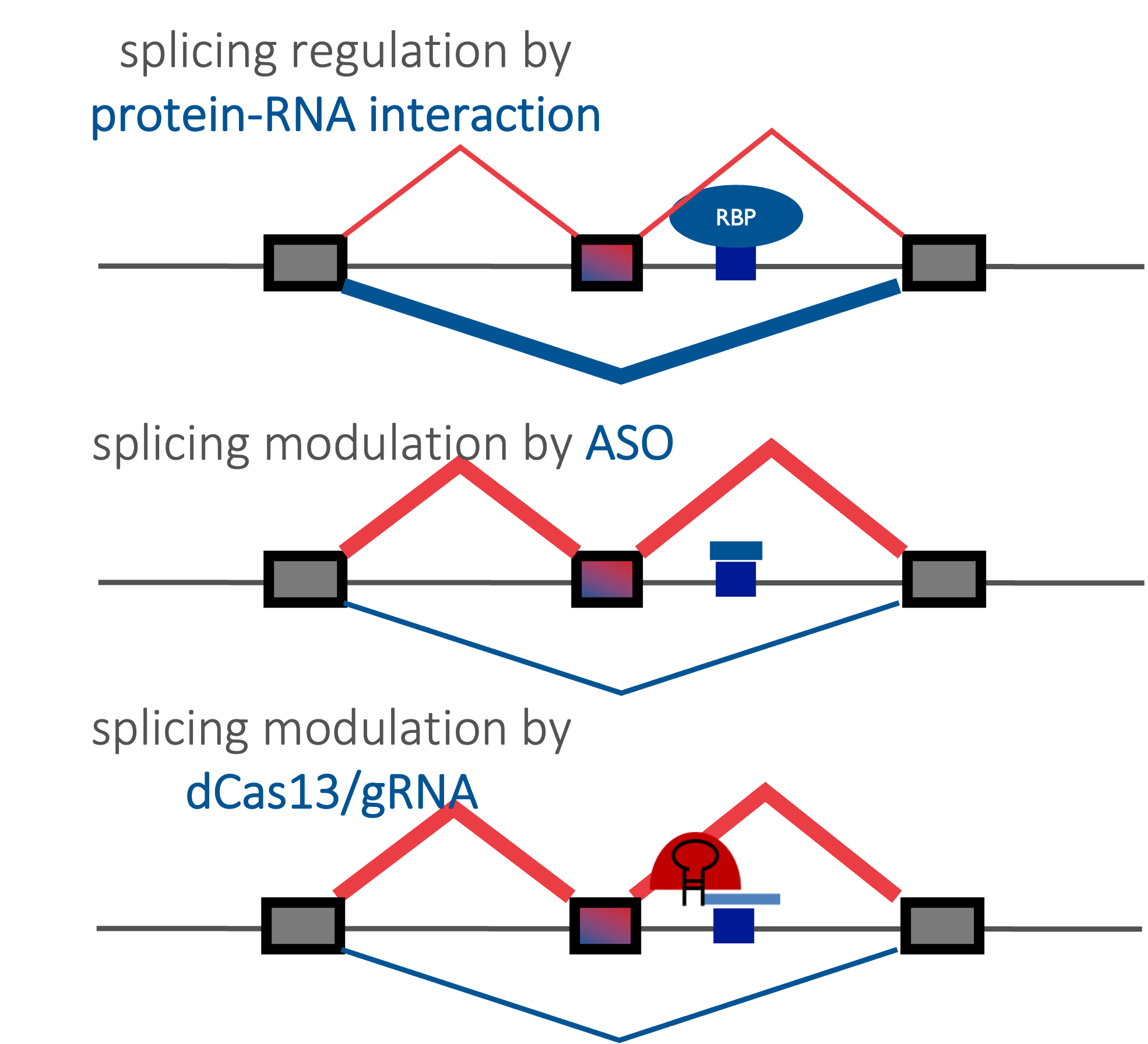
An ultimate goal of our research is to translate our knowledge of RNA regulation to RNA-based drugs. We are particularly interested in antisense oligonucleotides (ASOs) which has arisen as a new modality of drugs with massive potential for precision medicine. An ASO is a short stretch (15-30nt) of chemically modified DNA or RNA nucleotides that can be delivered to patients and bind to target RNA with high specificity and thereby modulate gene expression. Leveraging our unique target discovery platforms, we are working on specific treatment of several devastating neurologic and developmental disorders with tremendous unmet medical needs. We are also developing platform technologies (e.g., based on CRISPR) to speed up ASO screening, a bottleneck in the field.
|
 |
Protein-RNA interactions at single nucleotide resolution
|
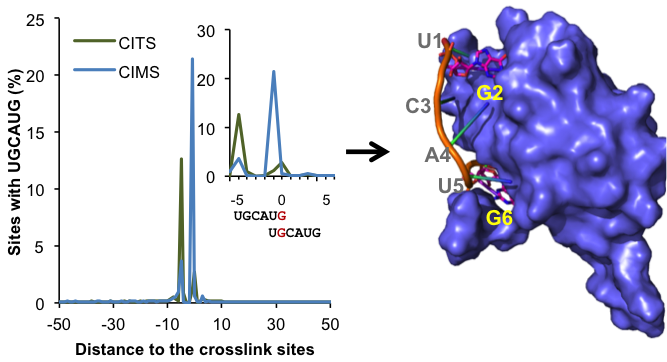
Post-transcriptional regulation of RNA processing such as alternative splicing is dictated by interaction of numerous RNA-binding proteins (RBPs) with their target transcripts. The first step to understand the function of RNA-regulatory networks is thus to precisely map protein-RNA interaction sites, which is challenging because most RBPs recognize very short and generate sequence motifs. The development of HITS-CLIP (or CLIP-Seq) has been a revolution to this field. We took advantage of HITS-CLIP data and developed crosslink-induced mutation sites (CIMS) and truncation site (CITS) analysis to map in vivo protein-RNA interactions at single nucleotide resolution on a genome-wide scale. Leveraging these maps of >100 RBPs using ENCODE and other datasets, we developed probabilistic models to learn subtle and quantitative rules of RBP specificity, which revealed novel modes of protein-RNA interactions and RNA regulation. We also mapped the first sets of allele-specific protein-RNA interaction sites with experimental evidence in the human transcriptome.
Software:
|
 |
Organizational principles and functional impact of neuronal RNA-regulatory networks
|
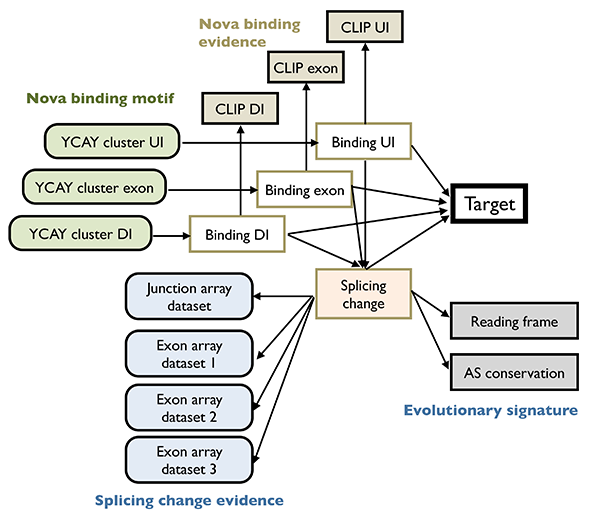
To understand the function of RNA regulatory network, a first step is to infer the structure of such networks, which is challenging due in part to the degeneracy and dynamic nature of the splicing code. We pioneered the development of integrative analysis of splicing regulatory networks, which has enabled us to identify alternative exons regulated by specific RBPs with unprecedented accuracy and sensitivity. In brief, our strategy was to identify altered splicing upon genetic RBP depletion, although such changes can be either direct or indirect. The latter was distinguished by mapping the precise RBP binding sites in vivo using HITS-CLIP. These data were then formally combined using a Bayesian network to determine high-confidence, direct target transcripts25,28. Studies using this paradigm demonstrated the concerted regulation of hundreds of alternative exons by individual neuronal RBPs. Investigation of the resulting networks allow us to make unexpected findings such as coupling of splicing with post-translational modifications.
|
 |
RNA regulatory networks in neural development and neuronal cell type diversity
|
The cellular diversity and functional complexity of the nervous system is derived from a tightly regulated developmental process, as dictated by precise molecular programs. Using an integrative approach, we investigated the contribution of splicing regulation to the transcriptome diversity and neuronal function during neurodevelopment using in vitro and in vivo systems. Our studies revealed the precise timing of developmental splicing switches as regulated by distinct combinations of RBPs and intriguing differences between CNS neurons and peripheral sensory neurons. Among them, we demonstrated that Rbfox is pivotal to establish the mature splicing program and its depletion in developing neurons impairs neuronal excitability due to abnormal axon initial segment formation. Our recent work also uncovered a combinatorial regulatory code that differentiate glutamatergic and GABAergic neurons in the cortex.
|
 |
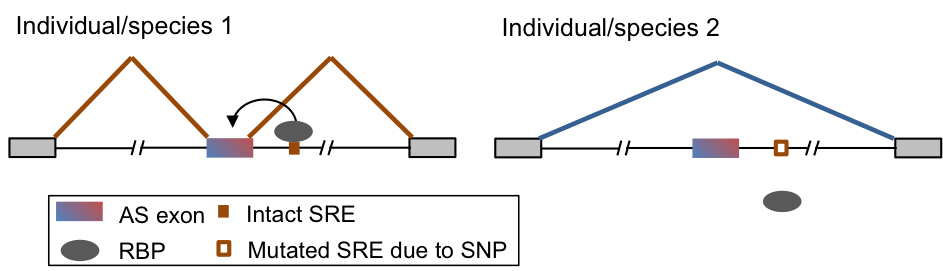
Variation of RNA-regulatory networks in evolution, human populations and in neuronal disorders
|
Another related direction of our lab is to evaluate the impact of mutations on RNA regulation in normal physiology or disease. Our study spans three contexts: comparison of different species (e.g. rodents and primates), different human populations, and patients affected by neurological diseases and normal controls. Such study is facilitated by our ability to determine protein-RNA interactions at a high resolution and distinction of functional vs. nonfunctional interactions. We are applying this strategy to parallel systems in different species that are directly comparable, large transcriptome profiles of human populations generated by consortium efforts, and mutations identified by genomic sequencing compiled from the public domain and collaborators.
|
 |
High-throughput transcriptomic data analysis
|
Our work heavily relies on high-throughput technologies which produce enormous amount of data, and on algorithms to transform these data into useful information. We are interested in developing better algorithms to process transcroptomic data, such as mapping RNA-Seq reads, discovering and quantifying RNA processing in specific conditions, and modeling the specificity of protein-RNA interactions.
|
 |